
Олефинирование по Джулия-Литгоу представляет собой многоступенчатый процесс синтеза алкенов, основанный на восстановительном элиминировании β-ацилоксисульфонов, образующихся при добавлении α-металлированных алкиларилсульфонов к карбонильным соединениям. Реакция была открыта Марком Джулия и Жан-Марком Пэрисоном в 1973 году, а впоследствии она была развита в более общую и полезную модификацию Коциенского и Литгоу.
Реакция протекает в четыре стадии:
- Металлирование арилалкилсульфона;
- Добавление образующихся карбанионов к альдегиду или кетону;
- O-ацилирование (или сульфонилирование) аддукта присоединения;
- Восстановительное элиминирование предпоследнего промежуточного интермедиата β-ацилоксисульфона.

Хотя все стадии можно проводить в одном реакционном сосуде, на практике более высокий общий выход обычно получается, если предпоследнее промежуточное соединение изолированно перед стадией восстановительного элиминирования. Аддукт присоединения 3 обычно образуется как смесь всех возможных диастереоизомеров; однако это не имеет никакого значения, потому что стереохимия в соединении 3 или 4, теряется на этапе восстановительного элиминирования. Отличительной чертой олефинирования по Джулия—Литгоу является его высокая транс-стереоселективность, являющаяся частью различных радикальных механизмов, которые действуют на последней стадии.
Наиболее распространенным вариантом олефинирования Джулия остается тот, который был предложен Коциенским и Литгоу, включающий в себя добавление литированных фенилсульфонов к карбонильному компоненту с последующим бензоилированием и затем восстановительным элиминированием, опосредованным 6% амальгамой натрия в системе растворителей MeOH-THF с фосфатным буфером. Стадия ацилирования снижает вероятность повторного ретроприсоединения, которое может происходить во время восстановительного элиминирования, но иногда ее можно пропустить без ущерба для общего выхода:

Факторы, влияющие на эффективность реакции и стереоселективность
Успешный результат олефинирования по Джулия—Литгоу зависит от правильной работы каждой из четырех стадий. Проблемы на стадии ацилирования возникают редко; однако природа компонентов, которые должны быть связаны, и используемые условия реакции являются решающими факторами, определяющими эффективность всех других стадий и ожидаемой степени стереоселективности и конечного выхода.
- Сульфоновое металлирование и присоединение к альдегидам и кетонам
α-протоны алкилфенилсульфонов имеют кислотность, сравнимую с кислотностью сложных эфиров (pK ~ 30 в DMSO), и депротонирование может быть достигнуто с помощью n-BuLi в растворителе THF при низкой температуре. В качестве альтернативы, если сульфон содержит электрофильные центры, которые могут быть атакованы алкиллитиевым реагентом, можно использовать ненуклеофильное основание, такое как LDA (диизопропиламид лития). Литированные первичные алкилфенилсульфоны имеют хорошую нуклеофильность и обычно добавляются к альдегидам для получения ожидаемых промежуточных β-алкоксисульфонов с превосходным выходом; однако их внутренняя основность может вызвать проблемы с легко поддающимися енолизации альдегидами. Однако превращение лития в менее основное металлоорганическое производное решает эту проблему.
Например, литий производное сульфона 8 не смогло присоединиться к α-окси альдегиду 12 во всех исследованных условиях, но трансметаллирование в реактив Гриньяра 10 или трифторборатный комплекс 11 привело к желаемому аддукту присоединения 13 (Рисунок 3). Последующее восстановительное элиминирование (без ацилирования спирта) дало E-алкен 14, с общим выходом более 80%.

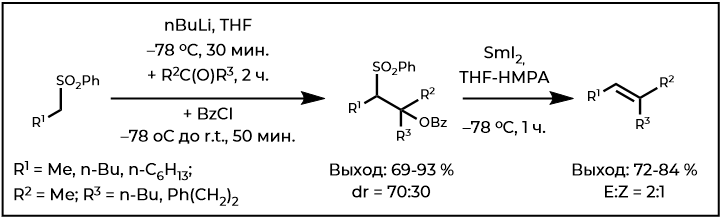
В случае же тризамещённых олефинов обязательно использовать либо вторичный алкилметаллированный сульфон, либо кетон. Оба класса соелинений имеют проблемы, которые могут привести к низкому выходу. Добавление первичных алкилметаллированных сульфонов к кетонам нежелательно в виду восстановления аддуктов β-алкоксисульфона в исходные продукты. И наоборот, перенос протонов может доминировать в попытках добавления стерически затрудненных (и более простых) вторичных алкильных металолитных сульфонов к енолизируемым альдегидам. В свою очередь, использование DME в качестве растворителя уменьшает степень енолизации. Другой же метод заключается в улавливании in situ промежуточного β-алкоксисульфона при низкой температуре BzCl или TMSCl. Использование BzCl в сочетании с SmI2 обеспечивает синтез тризамещённых олефинов с высоким выходом, но и с низкой стереоселективностью:
- Правила восстановительного элиминирования и влияние разветвления цепи на транс-селективность
Заметное улучшение транс-селективности отмечается для образования 1,2-дизамещенных алкенов путем восстановительного элиминирования β-ацилоксисульфонов, когда элементы разветвления цепи вводятся в заместители, которые будут «прикрывать» новую двойную связь. (Рисунок 5).
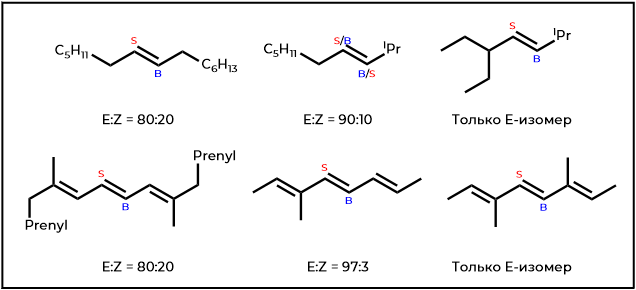
(S = исходный сайт группы PhSO2, B = исходный сайт группы BzO)
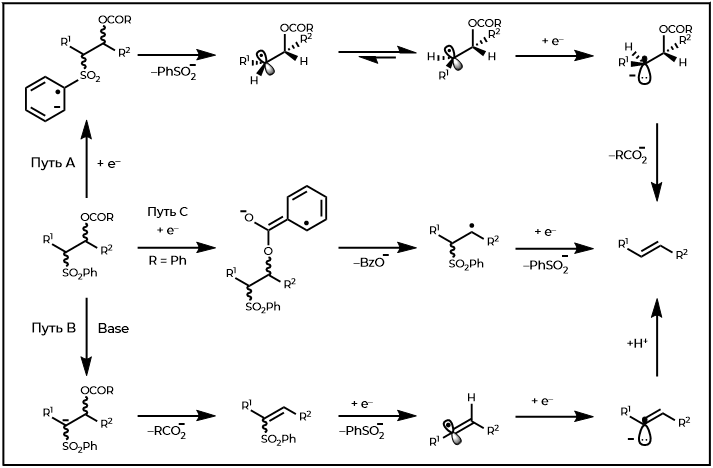
Механизм, объясняющий это поведение (обнаружен как для несопряженных, так и для сопряженных алкеновых продуктов), а также тот факт, что соотношение E:Z конечного продукта не зависит от диастереомерного состава исходного β-ацилоксисульфона, был сформулирован Коциенским и Литгоу (Путь А, Рисунок 6). Внедрение одного электрона в фенилсульфон с последующей потерей сульфината, как предполагается, приводит к радикальному промежуточному соединению, которое конформационно изменяется перед добавлением второго электрона и отщеплением кислородного нуклеофуга от зарождающегося карбаниона. Более поздние исследования поставили под сомнение достоверность этого механизма восстановления, опосредованного Na(Hg), но подтвердили его вероятную применимость в реакциях с использованием Sml2 в качестве альтернативного источника электронов. Кек выдвинул вторую механистическую гипотезу, чтобы лучше объяснить восстановление Na(Hg) (Путь B), а Марко позже предложил еще одну отличную возможность, которая потенциально применима к восстановлению вне β-бензоилоксисульфонов (Путь C).
Примеры реакций


Если вы заинтересовались этой реакцией, то можете почитать Comprehensive Organic Synthesis за авторством Paul Knochel и Gary A Molander.
Любите химию!